Understand the principle behind complex systems

Aoraki, NZ, 2019; photo by my wife
Feng Bao
I am a Postdoc working at Altschuler and Wu lab, University of California, San Francisco. I obtained my PhD from Tsinghua University in 2019. My research involves general learning approaches and their applications to biological and medical studies as well as drug screens.
Contact: feng.bao with the suffix @ucsf.edu
Research interests
- Recent focus on statistical learning
- Architectures to disentangle information within multi-modal/-omics inputs
- Variational information theoretic methods
- Constrastive learning for multimodal data
- Recent focus on biological questions
- Quantify interactions across multiple single-cell modalities
- Improve throughput of spatially resolved transcriptomics statistically
- Integrate high-content phenotypic drug profiles
Full publication list on Google Scholar
Recent works
In progress
- Integration of high-content imaging-based drug screens.
- Hypioxia multiomics.
- Spatial multiomics.
Enhancing the spatial resolution of diverse spatial omics platforms by joint generative modeling of space, image and omics
Tissue image (e.g., H&E) is always available along with the generation of spatial omics data. A typical tissue image can be in extremely high resolution (usually at least millions of pixels, note 1 million is just 1000 x 1000 pixels, smaller than the resolution of an iPhone photo) compared with the spatial omics resolution (usually in thousands). A natual idea is to use the high-resolution image to guide the enhancement of low-resolution omics. We tested this idea through a generative modeling of spatial coordinates, image and omics. It works for different omics types, spatial technologies and the recently emerging spatial multiomics. The work was published in Nature Communications.
Multi-modality structured embedding for spatially resolved transcriptomics analysis
Decomposing cell heterogeneity of complex biological systems is an important step to the comprehensive understanding of their organizations and mechanisms. Transcrptomics and imaging are two most widely used approaches to study tissue heterogeneity. Here we try to combine information from these two modalities and provide more extensive dissection of subpopulations in tissues. We follow two principles in design: (1) structured information from single modality (e.g. apparent subpopulations) are preserved after combinations; (2) corrupted information in one modality will not pollute the others.
[Project Page] [Code] [Publication]
Characterizing tissue composition through combined analysis of morphologies and transcriptional states. Feng Bao*, Yue Deng*, Sen Wan, Bo Wang, Qionghai Dai#, Steven J. Altschuler#, Lani F. Wu#. Nature Biotechnology. https://doi.org/10.1038/s41587-022-01251-z
Deep association kernel learning to explain the genetic causality for complex diseases
 Causal loci contribute to complex diseases in various manners. The comprehensive identification of suspicious genes requires a general genome-wide association study (GWAS) model that can work with different types of genetic effects. Here, we try to use a trainable framework to automatically detect these associated positions meanwhile provide a statistical significance quantification. This work was published as a cover paper in the new Machine Learning Journal of Cell Press Patterns.
Causal loci contribute to complex diseases in various manners. The comprehensive identification of suspicious genes requires a general genome-wide association study (GWAS) model that can work with different types of genetic effects. Here, we try to use a trainable framework to automatically detect these associated positions meanwhile provide a statistical significance quantification. This work was published as a cover paper in the new Machine Learning Journal of Cell Press Patterns.
[Project Page] [Code] [Publication]
Explaining the Genetic Causality for Complex Phenotype via Deep Association Kernel Learning. Bao, Feng, et al. Patterns 1.6 (2020): 100057.
Recurrent neural network for single-cell RNAseq imputations
scScope is a deep-learning based approach that can accurately and rapidly identify cell-type composition and transcriptional state from noisy single-cell gene-expression profiles containing dropout events and scale to millions of cells. This work was published in Nature Methods.
[Project Page] [Code] [Publication]
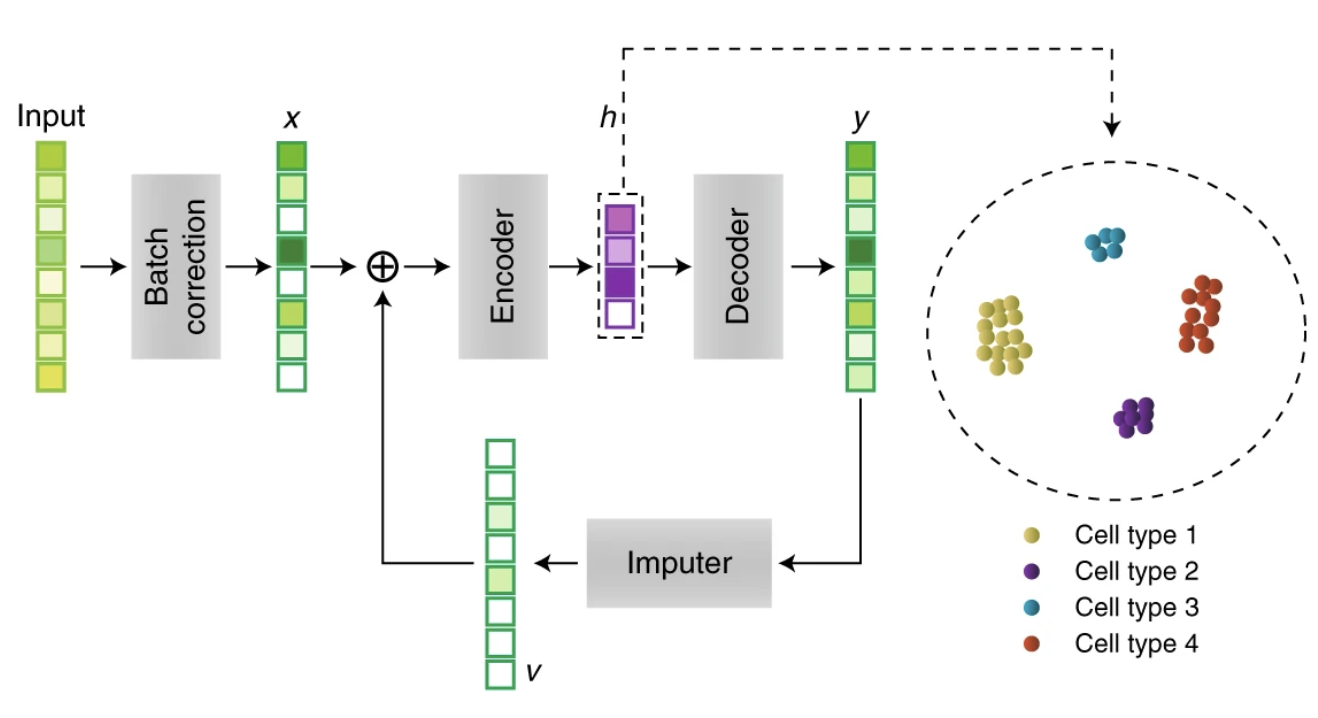
Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning. Yue Deng*, Feng Bao*, Qionghai Dai, Lani F. Wu#, Steven J. Altschuler#, Nature Methods, 2019, DOI: https://doi.org/10.1038/s41592-019-0353-7
Academic service
Reviewer for the following journals:
- Cell (but the editor seemed to forget my comments)
- Nature Biotech.
- Nature Biomedical Eng.
- Cell Systems
- Nature Communications
- Genome Biology
- IEEE Transactions on Fuzzy Systems
- IEEE Transactions on Neural Networks and Learning Systems
- Briefings in Bioinformatics
- IEEE Journal of Selected Topics in Signal Processing
last update: Jul 31st, 2024